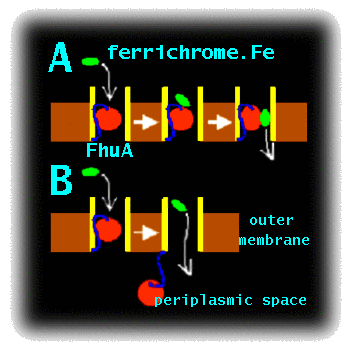
Project DescriptionMCFI-2000-01210 : Simulation Studies of the Mechanism of Bacterial Transport ProteinsCurrent work on this topic.To enter Gram negative bacteria solutes must cross two membranes. Ligands first must be transported across the outer membrane into the periplasmic space, and then must cross the inner membrane to enter the cytoplasm. Proteins of the outer membrane are particularly amenable to structure determination by X-ray crystallography. E. coli accumulates Fe3+ via transport of a siderophore - a small cyclic peptide or related molecule which chelates the ferric ion and retains it in solution. FhuA, the receptor for ferrichrome-iron in E. coli, mediates the active transport of ferric siderophores through the outer membrane of Gram-negative bacteria (Sansom 1999). It is representative of a wider family of bacterial outer membrane active transporters. Such transporters are possible targets for novel anti-bacterial drugs, as inhibition of transport could be used to impede bacterial growth. However, before such drugs can be developed, the mechanism of transport must be understood. Although X-ray diffraction studies have provided a high resolution structure of FhuA (Ferguson et al. 1998, Lochner et al. 1998), with and without its bound ligand (the Fe.siderophore complex), alternative approaches are needed to probe the conformational dynamics of this molecule. These include molecular dynamics (MD) simulations of the FhuA protein embedded in a fully solvated lipid bilayer. The FhuA protein consists of a hollow, 22-stranded, antiparallel beta-barrel, which is occluded by an N-terminal "plug" domain. X-ray crystallographic studies of the liganded and unliganded structures of FhuA reveal conformational changes affecting the plug, induced upon binding of the solute to the extracellular side. In particular, ligand binding results in unfolding of helix H1 of the plug, which is thought to trigger a subsequent interaction of FhuA with the energy transducing TonB complex. In addition, it has been proposed that a rearrangement of loops of the inner domain could lead to the formation of a narrow water-filled diffusion channel (a "protopore") that might allow the ligand to permeate. Alternatively, ligand transport might be associated with removal of the "plug" from the beta-barrel. The aim of this proposal is to use state of the art computer simulations to analyse the energetics and dynamics of these two proposed mechanisms (Fig. 1), in order to decide which is the more feasible.
Figure 1 : Cartoon of two possible mechanisms of transport by FhuA: A) Protopore and B) Plug Removal. This work will extend the current state-of-the-art in membrane simulations. To date, large scale (nanosecond) MD simulations have been performed on bacterial porins (Tieleman and Berendsen 1998) and on bacterial ion channels (Shrivastava and Sansom 2000), but have yet to be extended to more complex transporter proteins. However, preliminary simulations (Faraldo-Gomez and Sansom, unpublished results) on FhuA suggest that such simulations, though computationally challenging, are feasible. In addition to simulations on membrane systems, studies in a number of laboratories (e.g. Blondel et al. 1999) have demonstrated that computational approaches to search for reaction pathways and transition states can be successfully applied to complex biological systems. We intend to combine these two approaches in the proposed study of the FhuA mechanism. Research Project.The initial studies will extend our pilot MD simulations of FhuA in a lipid bilayer (Fig. 2) to check that the simulation system is stable over a multi-nanosecond timescale. In particular, we will address the question of the optimal simulation protocol, investigating the effects of e.g. adjustment of sidechain ionisation states to match predicted pKA values, and cut-offs vs. particle mesh Ewald methods for treatment of long-range electrostatic interactions. The optimal simulation protocol will be defined as that which best reproduces the loop mobilities as indicated by comparison of simulation RMS fluctuations and X-ray B-values.
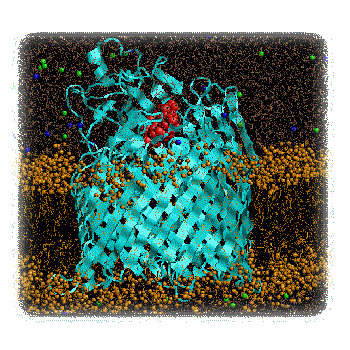
Figure 2 : FhuA (blue) plus Ferrichrome (red) in a DMPC Bilayer (brown). Pilot simulations (Faraldo-Gomez & Sansom, unpublished results). Having established an optimal simulation protocol, long (ca. 20 ns) MD simulations will be run of both the unliganded and liganded FhuA molecules, embedded in a phospholipid bilayer. Detailed analysis of the "penetration" of water molecules into the structure will reveal whether ligand-binding appears to unmask formation of a "protopore". Geometric (Connolly surface) calculations on simulation snapshots will be used to establish the dimensions of any protopore. "Steered" molecular dynamics simulations employ a standard MD potential function, with the addition of an external force applied to certain atoms. Such simulations may be used to dock/undock a ligand into a binding site, although one must take care not to interpret the results in terms of a pathway to/from a genuine transition state. We will employ such steered MD calculations in two modes to provide preliminary descriptions of the two models of the FhuA mechanism. To test the "protopore" mechanism, an external force will be applied to the centre-of-mass of the ferrichrome in order to "drag" it through the protopore(s) revealed by the water-penetration analysis. The energetics of ferrichrome/protein interactions and the degree of distortion of the FhuA as the ligand is dragged through it will be analysed. To test the "plug removal" mechanism, a force will be applied to the centre-of-mass of the N-terminal plug domain in order to extract it from the surrounding beta-barrel. Again, analysis will focus on the energetics of the process and the degree of distortion of the protein. Having compared the two mechanisms by steered MD simulations, and thus having established first approximations to reaction pathways for the two mechanisms, it will be possible to compare the two mechanisms in more detail by refinement of the proposed reaction pathways, and measurement of their relative transition state energies. In this way the more kinetically favoured pathway will be revealed. This analysis may be carried out using e.g. a similar method to that employed by (Blondel et al. 1999) in their analysis of the retinoic acid receptor. Our system will be a little larger as it will be necessary to include at least some explicit water molecules. Expected Results.The anticipated results of this project are a detailed energetic and structural comparison of two possible mechanisms for FhuA mediated transport. These will be important in terms of relating the structure of FhuA to ongoing experimental studies, in a number of laboratories, of the role of TonB in providing energy coupling to transport. Furthermore, our results will provide a paradigm for computational studies of other transporters as their structures emerge. Such studies will be of central importance in understanding the molecular basis of bacterial transport. As the outer membrane forms the "interface" of the bacterium with its external environment, it is an important potential target for novel anti-bacterial agents. This is clearly an area of considerable biomedical importance given the continuing war against bacterial infection and the development of resistance to many current antibiotics. Further links.
References.Blondel, A., J. P. Renaud, S. Fischer, D. Moras, and M. Karplus. 1999. Retinoic acid receptor: A simulation analysis of retinoic acid binding and the resulting conformational changes. J. Mol. Biol.291:101-115. Buchanan, S. K., B. S. Smith, L. Venkatramani, D. Xia, L. Essar, M. Palnitkar, R. Chakraborty, D. van der helm, and J. Deisenhofer. 1999. Crystal structure of the outer membrane active transporter FepA from Escherichia coli. Nature Struct. Biol.6:56-63. Ferguson, A. D., E. Hofmann, J. W. Coulton, K. Diederichs, and W. Welte. 1998. Siderophore-mediated iron transport: crystal structure of FhuA with bound lipopolysaccharide. Science.282:2215-2220. Lochner, K. P., B. Rees, R. Koebnik, A. Mitschler, L. Moulinier, J. Rosenbusch, and D. Moras. 1998. Transmembrane signalling across the ligand-gated FhuA receptor; crystal structures of free and ferrichrome-bound states reveal allosteric changes. Cell.95:771-778. Sansom, M. S. P. 1999. Membrane proteins: a tale of barrels and corks. Curr. Biol.9:R254-R257. Shrivastava, I. H., and M. S. P. Sansom. 2000. Simulations of ion permeation through a potassium channel: molecular dynamics of KcsA in a phospholipid bilayer. Biophys. J.78:557-570. Tieleman, D. P., and H. J. C. Berendsen. 1998. A molecular dynamics study of the pores formed by Escherichia coli OmpF porin in a fully hydrated palmitoyloleoylphosphatidylcholine bilayer. Biophys. J.74:2786-2801. |
|||
|
|





| $Date: 2006/07/26 22:17:44 $ | Home | LabPage | About... | Send feedback |
| Copyright © 2001-2007 Marc Baaden. | ||
 This work is licensed under a Creative Commons License. |
||