 | News | Research | Recherche (fr) | Project | Team members | YouTube channel | ...
| News | Research | Recherche (fr) | Project | Team members | YouTube channel | ...Axes | Extraction Liqu.Liqu. | Structures | Systèmes complexes (shown) | Développement | Publications | HDR
Simulation de systèmes biologiques complexes
Les protéines membranaires font parti des systèmes biologiques complexes auxquels je m'intéresse. Elles sont de première importance en biologie cellulaire et interviennent par exemple comme canaux ioniques, récepteurs de médicaments et transporteurs de solutés. En effet il a été estimé que 30% des gènes encodent des protéines membranaires, et que celles-ci représenteront la moitié des cibles potentielles pour le développement de nouveaux médicaments. Néanmoins il n'existe que très peu d'information structurale sur ces protéines. A ce jour on connait les structures atomiques de 111 protéines membranaires alors qu'il en existe des milliers.
Un axe de recherche abordé pendant mon stage post-doctoral concerne les protéines membranaires sous forme de tonneaux b présentes dans la membrane externe des bactéries gram négatives et qui sont encore peu étudiées par rapport aux assemblages d'hélices a plus communément observés pour les autres protéines membranaires.
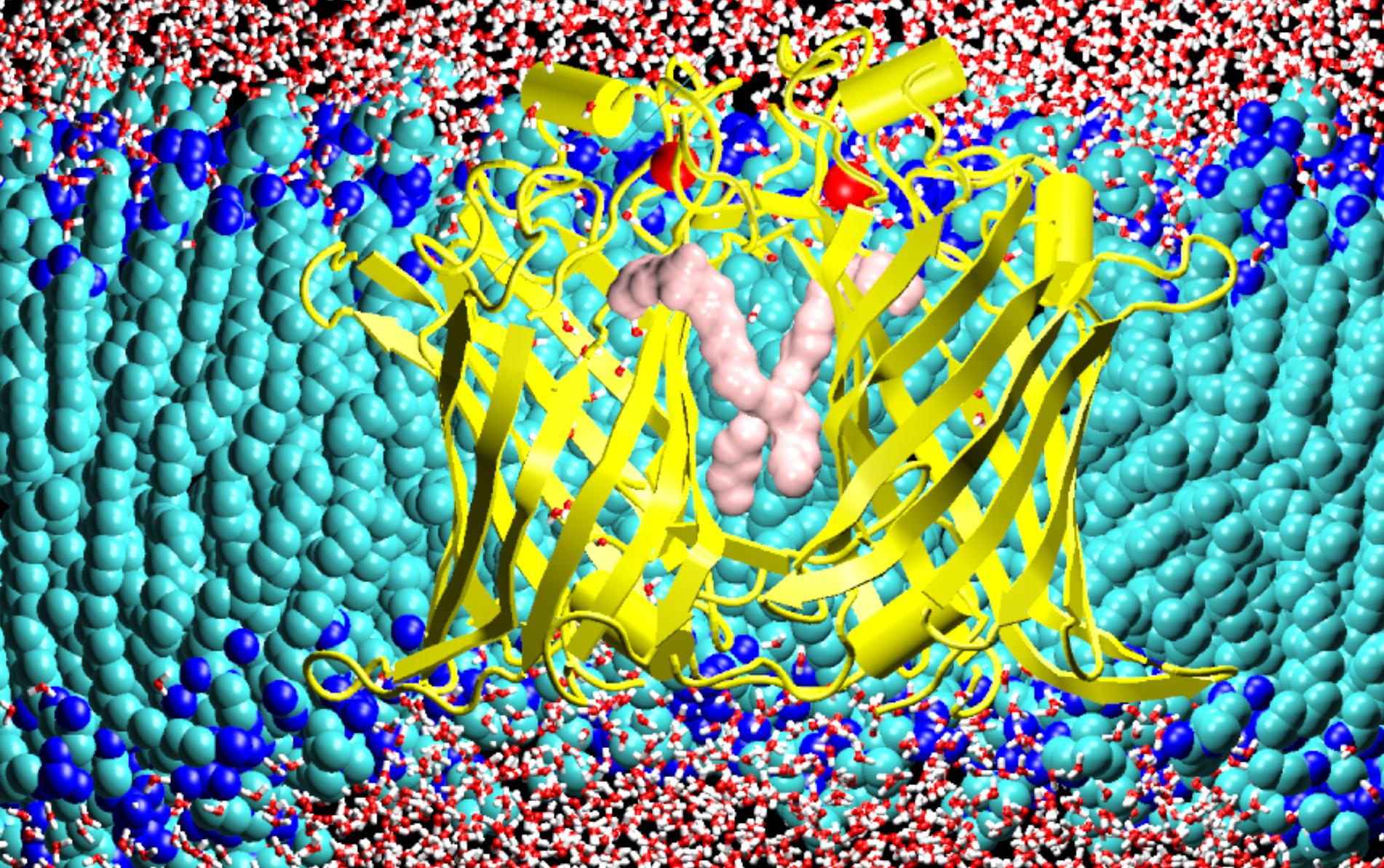
Le projet le plus ambitieux entrepris au sein des travaux sur les protéines membranaires concerne le transport de fer à travers la membrane externe des bactéries telles que E. coli. Les structures tridimensionnelles de plusieurs transporteurs du Fer, FhuA, FepA, FecA, et la structure du transporteur de vitamine B12 BtuB ont été résolues récemment. Ces protéines d'environ 75 kDa se présentent sous forme de tonneaux b à 22 feuillets dont l'intérieur est “bouché” par le domaine N-terminal d'environ 150 acides aminés. Actuellement, une question reste en suspens: ce “bouchon” se détache-t-il lors du transport ou subit-il un changement conformationnel important ?
La dynamique moléculaire nous aide à comparer les structures apo et holo de ce système, et nous espérons pouvoir en tirer des conclusions par rapport à l'éventuel détachement du “bouchon”. Nous avons commencé par la modélisation de la protéine FepA en y ajoutant les parties manquantes dans la structure publiée. Des calculs électrostatiques nous permettent de considérer l'influence du pH et de déterminer l'état de protonation le plus probable de la protéine. Nous avons également modélisé la molécule entérobactine qui complexe le fer lors de son transport. De longues simulations de DM (45 ns) dans un environnement modèle d'une membrane biologique en détail atomique ont été entrepris pour le système apo et holo. L'analyse détaillée de ces simulations est actuellement en cours [[Baaden en cours I]].
Plusieurs protéines de la membrane externe bactérienne sont des enzymes. Mon premier contact avec un système enzymatique était l'étude de la protéine membranaire Ompla décrite ci-dessous. Depuis, des simulations sur l'enzyme OmpT ont été publiées [[Baaden 2004]]. Dans le cadre du projet BioSimGrid j'ai contribué de façon importante à la comparaison d'un ensemble de simulations sur 4 enzymes de type hydrolase dont Ompla et OmpT font partie. Une publication vient d'être soumise [[Baaden 2007]].
L'enzyme OmplA présente une architecture de tonneaux b et est d'une taille d'environ 33 kDa. Les fonctions exercées par cette protéine dans la membrane externe comprennent la stabilisation de la structure membranaire et l'activité enzymatique. Les structures de plusieurs formes fonctionnelles de cette enzyme ont été déterminées par cristallographie, mais n'ont pas permises d'élucider les relations entre structure et fonction. Pour obtenir des informations complémentaires, nous avons effectué des simulations de dynamique moléculaire dans une bicouche lipidique pour trois formes de cette enzyme: le monomère, inactif, le dimère sans substrat et le dimère en présence d'un inhibiteur. Nous avons montré que la présence de molécules d'eau bien spécifiques, d'ions calcium et la formation d'un réseau de liaisons hydrogène autour de ceux-ci jouent un rôle important dans le cycle enzymatique. De même la dynamique conformationnelle de la poche accueillant le substrat semble déterminant pour réguler l'activité de l'enzyme: en absence de substrat la poche se ferme et empêche l'accès au site actif. Ce travail interdisciplinaire a été publié dans un journal de biologie moléculaire [[Baaden 2003a]] et a été primé par le groupe de graphisme et modélisation moléculaire (GGMM) qui m'a attribué son prix de jeune chercheur lors de son congrès bi-annuel 2003 à Cabourg.
Les simulations des espèces Ompla et Fepa s'inscrivent dans une étude plus systématique de protéines sous forme de tonneaux b de taille variable. Pour compléter la gamme des protéines étudiées, j'ai dernièrement effectué des simulations sur l'enzyme OmpT qui est de plus petite taille. On peut ainsi comparer ces tonneaux à 10, 12 et 22 feuillets avec d'autres systèmes simulés.
Tous ces systèmes ont été simulés dans une bicouche lipidique entièrement hydratée, en variant les conditions de simulation par rapport aux interactions électrostatiques à longue distance ainsi que d'autres paramètres tels que le couplage de température et de pression. L'ensemble des simulations présente un travail systématique sans précédent. J'ai mis en évidence l'architecture du site actif et l'importance d'une molécule d'eau qui pourrait intervenir dans la réaction enzymatique. Un premier modèle approximatif d'un complexe enzyme-substrat issu de calculs d'arrimage moléculaire a été proposé et ce modèle a depuis été amélioré. Cette étude a également suscité des travaux par d'autres groupes, notamment chez P. Carloni en Italie.
Les résultats concernant l'influence des approximations et conditions de simulation sur les conclusions physico-chimiques et biologiques sont très précieux pour les travaux futurs où on sera amené à simplifier les modèles pour permettre un calcul en temps réel.
Une autre famille de protéines membranaires est celle des protéines de la famille dite ABC (ATP-Binding cassette) qui se chargent du contrôle de l'osmorégulation et de la détoxication des xénobiotiques tels que les métaux lourds, les agents pathogènes ou certains médicaments. Leur importance clinique et biologique est lié à leur capacité de conférer la résistance à divers traitements, notamment la chimiothérapie. Une seule mutation peut également entraîner des maladies tels que le diabète ou la mucoviscidose.
Dans un premier travail, nous avons proposé un modèle détaillé du transporteur ABC Msba qui peut servir de point de départ pour la modélisation par homologie d'autres protéines de la même famille qui sont des cibles thérapeutiques [[Baaden 2003b]]. Parmi mes publications c'est la plus citée à ce jour. Une deuxième structure à l'état solide obtenue très récemment confirme en grande partie notre modèle proposé.
Notre collaboration sur les transporteurs ABC se poursuit dans l'étude du complexe SUR/Kir qui est menée sur les fronts expérimentaux et théoriques simultanément [[Baaden en cours II]]. Ce complexe a un rôle de première importance dans le diabète et Frances Ashcroft qui est impliquée dans ce travail en est une experte mondialement reconnue.
[Baaden en cours I] M. Baaden et M.S.P. Sansom : "“In Silico Study of the Iron Transporter FepA and its Complex with Enterobactin”", en rédaction.
[Baaden 2004] M. Baaden et M.S.P. Sansom : "“OmpT: molecular dynamics simulations of an outer membrane enzyme”", Biophys.J. , 2004, 2942-2953.
[Baaden 2007] K. Tai, M. Baaden, S. Murdock, B. Wu, M.H. Ng, S. Johnston, R. Boardman, H. Fangohr, K. Cox, J.W. Essex et M.S.P. Sansom : "“Three hydrolases and a transferase: comparative analysis of active-site dynamics via the BioSimGrid database”", Biochemistry 2007, soumis.
[Baaden 2003a] M. Baaden, C. Meier et M.S.P. Sansom : "“A molecular dynamics investigation of mono- and dimeric states of the outer membrane enzyme Ompla”", J.Mol.Biol. , 2003, 177-189.
| $Date: 2011/01/12 22:51:43 $ | Home | LabPage | About... | Send feedback |
| Copyright © 2001-2007 Marc Baaden. | ||
 This work is licensed under a Creative Commons License. | ||