

Nico_chimera project
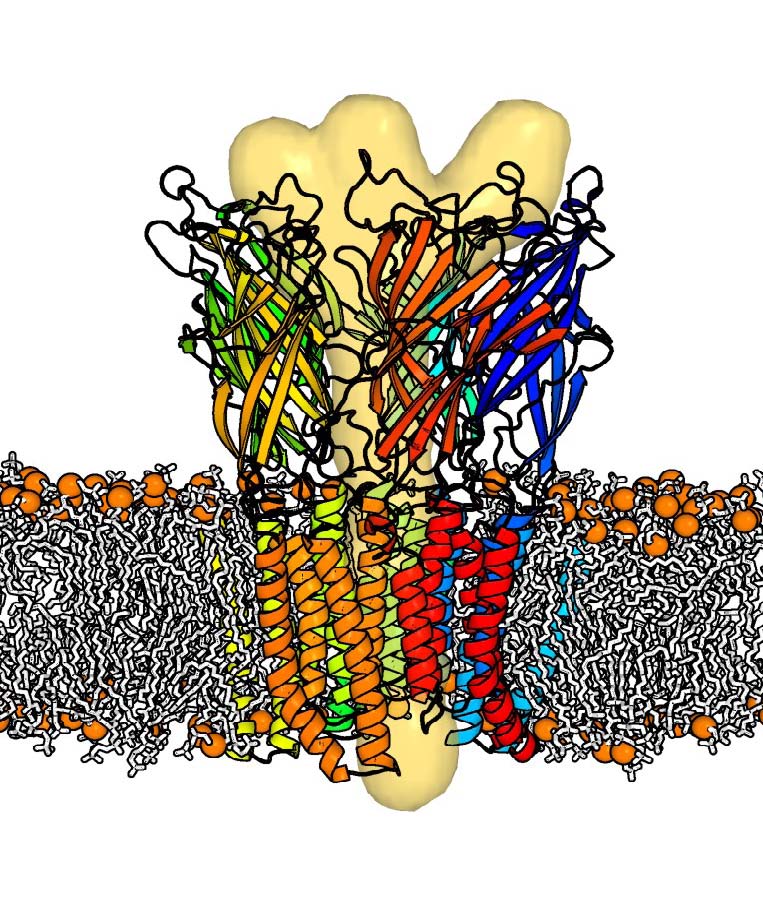
We have recently completed the structure determination at 2.9 Angstrom resolution of a bacterial homologue (GLIC) of the pentameric nicotinic acetylcholine receptor, a member of the CysLoop receptor (CLR) family. The gating of this ion channel is mediated by the binding of protons. Here we have crystallized the molecule at acidic pH, leading to an open conformation. Earlier in the same year, another member of the same family (ELIC) had been solved in a closed conformation. However, the sequence identity between the two molecules is too low to allow for a reliable homology modelling of the closed form of GLIC from ELIC (and vice versa). We have also undertaken molecular dynamics studies of ELIC in a fully solvated lipidic environment at neutral pH. A 1 micro-second long simulation revealed a quick closing of the pore channel, followed by sequential tertiary rearrangement of two consecutive subunits. We believe that this represents a significant part of the transition, but that more than half of the transition is still missing and currently out of reach of available computer power. We now want to attack the problem more directly by solving the crystal structures of the same cys-loop receptor in both open and closed forms. To this end, we will make heavy use of synthetic genes of chimera involving the combinatorial fusion of the extra-cellular domain (ECD) and trans-membrane domain (TMD) of various bacterial and eukaryotic genes. The design of such constructs will be aided by computing modelling using existing structures. In addition, we will work on the redesign of the agonist binding pocket by engineering the Loops A,B,C,D,E,F in the ECD. All constructions will be tested by electrophysiology using proteins expressed at the surface of oocytes. Those successful (i.e. active) constructs will be subjected to crystallisation, diffraction data collection and structure refinement when appropriate. We already have characterized such an active chimera and collected low resolution diffraction data for crystals of the apo form. In addition, we will try to crystallize binary complexes with both the agonist and inhibitors of the anesthetics type. We have already shown that some anesthetics modulate (inhibit) the opening of GLIC, and have determined the structures of 2 of these complexes at 3.2 Angstrom resolution. We are currently pursuing the interpretation of this structural data using our molecular dynamics trajectories. On the computational level, efforts to understand the electrostatics of this system upon pH change will be pursued using newly developed software. Finally, once we have the structures of the same molecule in the two extreme forms, we will simulate the transition path between the two forms using a coarse-grained method based on the maximum likelihood principle and an energy landscape for the two forms simplified by the so-called Elastic Network Model. We have recently improved this method to tackle very large molecules comprising as many as 15,000 atoms. In this manner, we should be in a position to predict in silico the effect of point mutations on the gating rate, shifting the protein engineering principles to the field of kinetic optimization. The most effective predicted mutants will be subjected to an experimental functional test.
See Marc Delarue's website (project coordinator) for more details.
This project has been selected by the ANR funding agency in the 2010 program.




